COOT Quick Reference Guide
Table of Contents:
Back to Facilities-Software Main Menu
Remarks
Our intention is not to provide
in-depth model building support here at SSRL, but to offer an easy-to-use graphics
program so that users who have collected data here can quickly look at their
maps. For example, if a MAD data set has
been collected and roughly phased using our solve script, the user may wish to
quickly check the quality of the resultant maps to determine whether additional
data should be collected to improve the solution. To this end, we have found the program COOT
to be very easy to run and inspect electron density maps with no prior
knowledge of the program.
Coot is molecular graphics program developed in York
and is used for model building, model completion and validation. It has some features that resemble those of
Turbo-Frodo, O, Quanta and XtalView's XFit, such as pull-down menus (see the image below), however
Coot does not do many aspects of structure representation. Coot displays maps and
models and allows model manipulations such as idealization, real space refinement,
manual rotation/translation, rigid-body fitting, ligand search, solvation, mutations,
rotamers, and Ramachandran plots. It also has a very robust superposition algorithm.
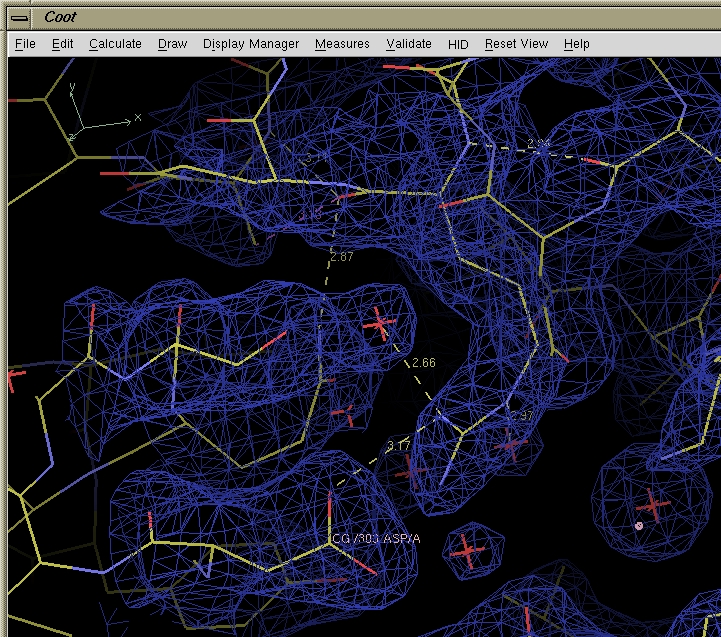
Screen capture from COOT showing a molecule and electron density.
Files, References and Documentation
Coot reads coordinate files in pdb format, and can also read pdb files which
have been compressed with gzip. Coot reads maps in CCP4 format (such as those generated with FFT). Coot is also able to
reads mtz files and can either calculate maps from data columns in the mtz
file specified by the user, or it can automatically generate maps from mtz files
containing map coefficients with column labels FWT, DELFWT, PHWT and PHDELWT (such as those produced by REFMAC).
Reference
The reference for the coot is:
Emsley P, Cowtan K (2004). Coot: model-building tools for molecular graphics.
Acta Crystallogr. D60, 2126-2132.
If using "SSM Superposition", please cite:
Krissinel E, Henrick K (2004). Secondary-structure matching (SSM), a new tool for
fast protein structure alignment in three dimensions
Acta Crystallogr. D60, 2256-2268.
Documentation
If you have detailed questions about these programs, please refer to the
COOT
documentation,
available on-line.
In order to get you quickly started
with Coot so that you can look at your MAD maps, here are some of the main
commands you might need to know. This is
not meant to be a detailed description of how to build you model or fit your
sequence to the electron density. If you
wish to continue using Coot once you leave SSRL we suggest that you download
your own copy, go through the manual and the
tutorial
and have fun!
Mouse functions
|
Left-mouse Drag
Ctrl Left-Mouse Drag
Shift Left-Mouse
Right-Mouse Drag
Ctrl Shift Right-Mouse Drag
Middle-mouse
Scroll-wheel Forward
Scroll-wheel Backward
Ctrl Right-Mouse Drag Up/Down
Ctrl Right-Mouse Drag Left/Right
|
Rotate view
Translates view
Label Atom
Zoom in and out
Rotate View around Screen Z axis
Centre on atom
Increase map contour level
Decrease map contour level
changes the slab (clipping planes)
translates the view in screen Z
|
If you don't have a wheel mouse (for example, on an SGI),
you can use the keyboard to increase and decrease the contour level as
described below.
Some Keyboard controls
You can find a great little
cheat-sheet here that you can
print out and keep handy .
|
Space
Shift-Space
N
M
D
F
F5
F6
F7
|
Next Residue
Previous Residue
Zoom out
Zoom in
Slim clip (slab less)
Fatten clip (slab more)
Open the Model/Fit/Refine dialog
Open the GoTo Atom Window
Open the Display Control Window
|
To increase or decrease contour
levels on maps, use + or -
on the keyboard. You need to make sure
you are controlling the correct map (in cases where you have more than one map
loaded). See the section on the Display
Manager below for details
Useful Menu Items
File
| open coordinates |
You can then browse for the pdb file you wish to load. |
| Auto Open MTZ |
You need to then specify a file which contains map coefficients with column
labels FWT, DEFWT, PHWT and PHDELWT (such as output by REFMAC). Coot will
automatically generate the Fo-Fc and 2Fo-Fc maps from this. |
| Open MTZ, CIF, phs etc |
You need to specify the file you wish to generate
a map from, and then select the correct column names for amplitudes and phases
(and weights if necessary). If your
phases have come from SOLVE/RESOLVE or Shelxe then you will have to use this option to
calculate your first Fo map. |
| Close molecule/map |
Used to delete a molecule or map from the display manager. |
| Save coordinates |
As the name implies, used to save any changes to a file. |
Calculate
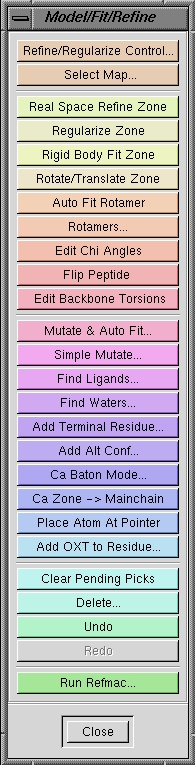
| Model/Fit/Refine |
This opens a floating menu (shown below) which contains all the
functions necessary for modeling
and refinement. |
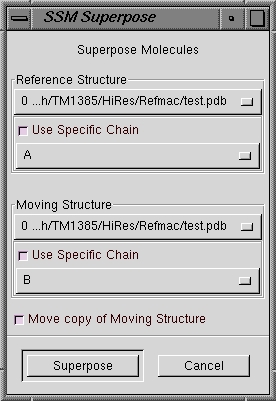
| SSM Superpose |
Starts the SSM superposition calculation and prompts for two models
to superimpose. |
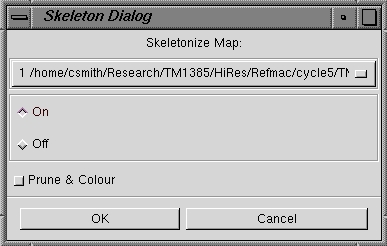
| Map Skeleton |
Starts a dialog box in which you can specify which map to skeletonize, normally
from a RESOLVE map there will be just one option, your initial Fo map.
You activate the skeleton by clicking the on
radio button and then OK. |
Draw
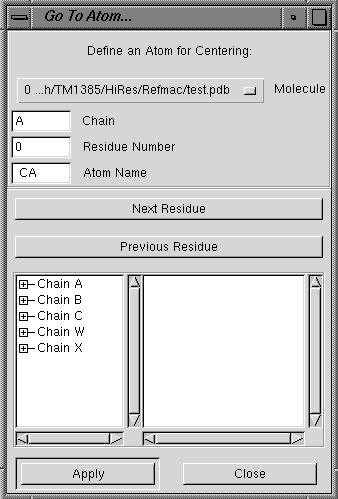
| Go To Atom |
Opens a dialog box (see below left) in which you can choose
the molecule you wish to work with, a chain name, residue number and atom
name. Alternatively, there is a listing
of the chains present in the map and these can be opened by clicking the +
button to the left of the chain name. The residue number can then be browsed from
the tree. To center on a particular residue, click that
residue number and then click the Apply button. You will, by default, be
centered on the CA atom. |
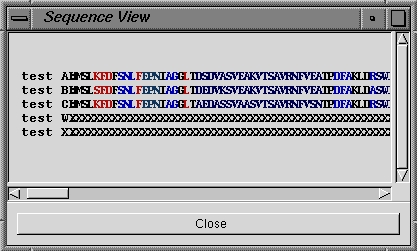
| Sequence View |
Selecting this option then allows you to browse a list of all the
molecules currently open. Selecting an
open molecule will then open a Sequence View dialog (see below right) with the sequences of
the chains listed in one-letter format. To center on a particular residue, click on the residue label and the
view on the screen automatically updates. |
| Cell & Symmetry |
Opens a Show Symmetry dialog which allows you to
select how you want to display the symmetry atoms. |
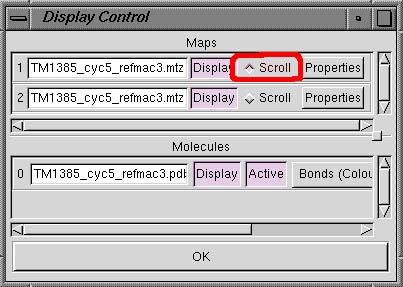
Display Manager
| Clicking this
option brings up the Display Control dialog box which lists the
currently loaded Maps and Molecules. You are then
able to toggle these various objects on and off using the Display button, change
some map features (eg color) using the Properties
button, or alter how you which to show the molecules (eg all atoms, CA only etc)
using the Bonds pull-down menu. In the Linux and SGI versions of Coot, there
is also a Scroll radio button beside each map name (see below). This button allows you to switch
between the different maps so that the sigma levels can be changed. |
Measures
| Distances & Angles> |
Distances and angles will be displayed on the screen and in the console
after mouse clicking on the atoms of interest. |
| Environment Distances |
Select the Show Residue Environment radio button and click OK.
Then use the middle mouse button to click on any
atom in a residue to see the contacts to all neighboring residues. |
Validate
| Ramachandran Plot |
This will give you a list of currently loaded molecules. Select the molecule
you wish to calculate the Ramachandran plot for. |
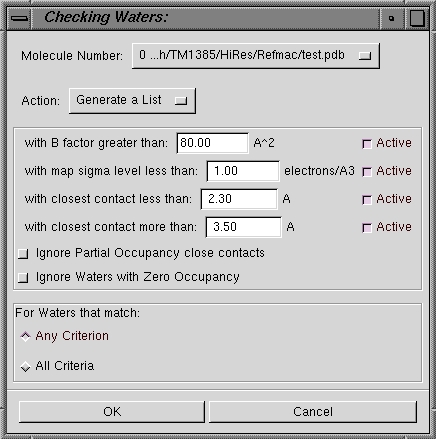
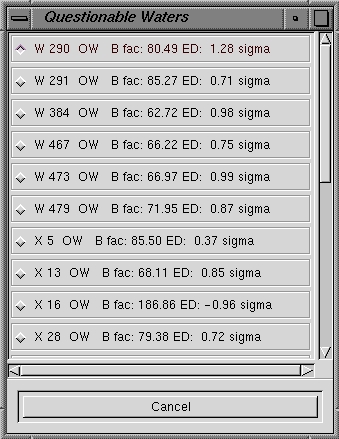
| Check/Delete Waters |
This dialog box (see below left) allows you to list or delete water molecules which
fall outside certain criteria. The
default action is to Generate a List but you can also Delete Waters
immediately if you so wish. If you
choose to list the waters (this is the default), a window will appear with the
water molecule names (below right). You can then work
your way through the list by clicking on the water molecule name and visually
inspecting each suspect water molecule. |
HID
| Scrollwheel |
Used to change which map is controlled by the scrollwheel
or the +/- keys if you are using keyboard contouring (in cases where more than
one map is loaded). |
|