areaimol - Analyse solvent accessible areas
areaimol XYZIN
foo_in.pdb
[XYZIN2
foo2_in.pdb]
[XYZOUT
foo_out.pdb]
[Keyworded input]
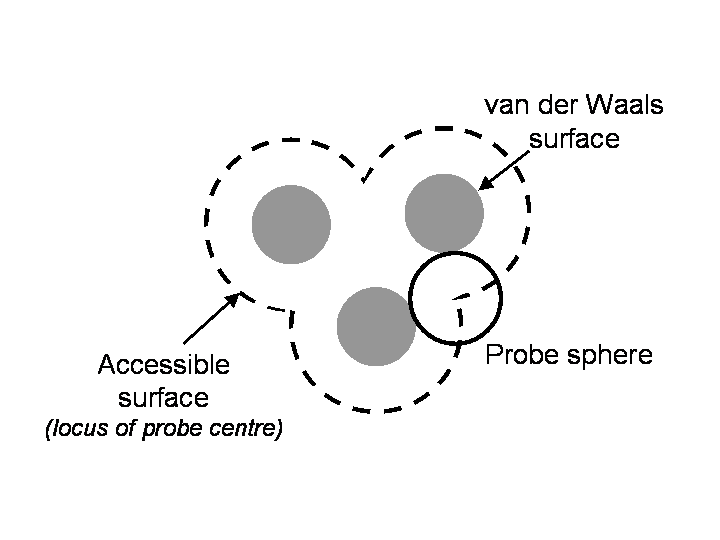
The solvent accessible surface of a protein is defined (Lee and Richards (1971)) as the locus of the centre of a probe sphere (representing a solvent molecule) as it rolls over the Van der Waals surface of the protein (figure 1). AREAIMOL calculates the solvent accessible surface area by generating surface points on an extended sphere about each atom (at a distance from the atom centre equal to the sum of the atom and probe radii), and eliminating those that lie within equivalent spheres associated with neighbouring atoms. This is different from the original Lee and Richards (1971) algorithm, which is implemented in the program SURFACE. Note also, that the solvent accessible surface is distinct from the molecular surface, which is the locus of the inward-facing point of the probe sphere (the sum of the contact and re-entrant surfaces - see figure 2).
AREAIMOL finds the solvent accessible area of atoms in a PDB coordinate file, and summarises the accessible area by residue, by chain and for the whole molecule. It will also calculate the contact area (the area in square angstroms on the Van der Waals surface of an atom that can be contacted by a sphere of the given probe radius) and will attempt to identify isolated areas of surface (which could be cavities either within the molecule, or formed as a result of intermolecular contacts).
It is capable of excluding specified residues from the calculations, and of generating symmetry related molecules. It can also be used to compare accessible area and analyse area differences. Accessible areas (or area differences) for individual atoms can be written to a pseudo-PDB output file.
This is an extensively revised version of the old AREAIMOL program which now also incorporates the functions of DIFFAREA, RESAREA and WATERAREA. The flexibility of the area calculation has been extended by the addition of new keywords PROBE (sets probe radius), PNTDEN (sets precision of area calculation) and ATOM (allows new atom types to be defined).
As of CCP4 version 6.1 the algorithm for calculating the accessible surface area has been made more accurate - see the Notes section.
The keywords are split into three groups:
This keyword controls the program function, the data required, and how it is processed and analysed (see PROGRAM FUNCTION). DIFFMODE must be the first keyword, unless it is omitted in which case the program defaults to DIFFMODE OFF.
Subkeywords:
- OFF [default]
- This corresponds to the function of the original AREAIMOL program. A single input file is required and a single accessible area calculation and analysis is performed.
- IMOL
- This mode analyses the differences in accessible areas of a molecule due to different intermolecular contacts (generated from different sets of symmetry operators and/or lattice translations).
- WATERS
- This mode analyses the differences in accessible area for the waters in a molecule depending on whether they are treated as solvent or as protein.
- COMPARE
- This mode is used to analyse the area differences for atoms and residues which are common between molecules held in two different files (XYZIN and XYZIN2).
The value of DIFFMODE may moderate the behaviour of other keywords: MODE, SMODE, SYMMETRY and TRANS (see below).
Controls which type of residues are included and how they are treated. There are four possible modes of operation, specified by one of the subkeywords below.
Subkeywords:
NOHOH [default] All waters (residue type HOH or WAT) are ignored. HOH The accessible area will only be calculated for waters (HOH or WAT), treating other waters as solvent. Only waters will be analysed. HOHALL As HOH, but waters are treated as protein, and consequently more waters will have low solvent accessibility. ALL Calculate accessible area for all atoms, including waters if present in file. Water atoms are treated as solvent when calculating accessible area.
Warning: waters may have large accessible area assigned in this MODE, leading to unrealistically inflated estimates of the total accessible area. Check the output carefully.
Under DIFFMODE WATERS, the MODE keyword is redundant and is ignored.
Symmetry mode keyword which is used to look at intermolecular contacts. There are two options:
Subkeywords:
- IMOL
- Account for intermolecular contacts by generating symmetry related molecules from coordinates in Brookhaven file before calculating accessible areas, using symmetry operators supplied by the SYMMETRY keyword. (The TRANS keyword can also be used, to generate molecules related by lattice translation symmetry.)
- OFF [default]
- Symmetry related atoms will not be generated and intermolecular contacts are not accounted for.
Under DIFFMODE IMOL, the SMODE keyword is redundant and is ignored.
Read the symmetry operations, specified as a name (e.g. P212121), the International Tables number, or as a series of symmetry operations (e.g. SYMMETRY X,Y,Z * -X,Y+1/2,-Z). In the latter case, all the symmetry operators must be supplied on a single SYMMETRY keyword.
If the SYMMETRY keyword is omitted when SMODE has been specified as IMOL then the program will generate symmetry related molecules assuming P1 symmetry (essentially, lattice translations only). If SMODE is OFF then the SYMMETRY keyword is optional.
Under DIFFMODE IMOL a second SYMMETRY keyword is necessary, to specify the symmetry operators required for the second area calculation (see below).
Note that unlike previous versions of the program, it is no longer necessary to manually exclude the identity operation when entering symmetry operations. The identity is implicitly assumed. If the identity is the only operation that has been entered (or if P1 symmetry is specified) then a warning may appear, but this can be ignored (unless you are not in P1 symmetry).
TRANSlation keyword. This causes the program to generate additional symmetry-related molecules by applying 125 translations made up from linear combinations of the primitive lattice vectors (+/-2 lattice vectors in each direction). Combining these with the spacegroup operators via the SYMMETRY keyword will generate the crystal lattice.
Only takes effect if DIFFMODE IMOL or SMODE IMOL have been specified.
Subkeywords for DIFFMODE IMOL:
- 1 (or 2)
- Apply the translation vectors on the first (or second) area calculation only.
- BOTH
- Apply the translations on both the first and the second area calculation.
- NONE
- [Default] Do not apply any translations.
For SMODE IMOL, NONE turns off the translations [default] and TRANS on its own is sufficient to switch them on.
Add or change an atom type and associated Van der Waals radius recognised by the program. <name> is the element name (as appears in columns 13-14 of the pdb file), and can be given in either upper or lower case (it is automatically upper-cased and right-justified before being processed). <no> is the atomic number and <radius> is the Van der Waals radius to be assigned to this atom type, in Angstroms.
If both <name> and <no> match those belonging to an atom already in the list then its Van der Waal radius will be changed to <radius>. If only one of either match, then the program ignores that occurrence of the ATOM keyword and the radius will remain unchanged.
AREAIMOL assumes a single radius for each element, and only recognises a limited number of different elements. Unknown atom types (i.e. those not in AREAIMOL's internal database) will be assigned the default radius of 1.8Å. The list of recognised atoms is:
Name Atomic no. VdW rad. (Å)
-----------------------------
C 6 1.80
N 7 1.65
O 8 1.60
MG 12 1.60
S 16 1.85
P 15 1.90
CL 17 1.80
CO 27 1.80
The ATOM keyword must appear once for each atom definition. The program can store up to twelve new atom types, in addition to those listed above.
Here residuen represents a three-character residue name (e.g. ARG for arginine). Atoms belonging to any of the named residues will be ignored in the area calculations, and will not be written to the output Brookhaven file.
Any number of specified residue names can appear together after a single EXCLUDE, separated by a space (e.g. EXCLUDE PRO ARG GLY). The EXCLUDE keyword can also be repeated any number of times with one or more specified residue names.
There is a maximum number of excluded residues which is set inside the program (currently 30). If there are more than this limit then extra names will not be recorded. Names entered in lower case will automatically be converted to uppercase. Note also that the program does not check that the entries given are valid residue names, or if any are repeated.
In DIFFMODE COMPARE, the named residues will be excluded from both of the input files before the areas are calculated.
Default: ALL
In DIFFMODE COMPARE MATCHUP sets the comparison criteria used when doing comparison of XYZIN and XYZIN2:
Atoms which are not included in the comparison are ignored in the output. MATCHUP is only available for DIFFMODE COMPARE.
The pointdensity keyword sets the precision of the area calculation. <point_density> is the number of points per square angstrom, so that the smallest area that can be calculated is the reciprocal of this value. The default is <point_density> = 15 points per square angstrom.
Note: High values of <point_density> allow more precise estimates of the accessible surface area, but will take longer to calculate - and if <point_density> is too large then the program may exceed its memory resources and stop. At lower values of <point_density> it is possible that atoms with low surface accessibility may be diagnosed as having no accessible surface area at all.
Sets the radius of the solvent molecule used as a probe in the area calculations to be equal to <x> angstroms.
The probe radius must be greater than zero, up to a limit of 25Å. The default radius is 1.4Å.
Switch on extended (i.e. verbose) printer output. In addition to the output described in 'PRINTER OUTPUT', the log file will also contain the following information:
The OUTPUT keyword causes a list of atoms to be written to the file with logical name XYZOUT. This file has a pseudo-PDB format and should contain the CRYST1 and SCALE cards from the input file, plus for each atom: the coordinates, the associated residue, and the accessible area (if DIFFMODE OFF) or area difference (in other DIFFMODES) in the B-factor column. This is intended to mimic the output from the old AREAIMOL program.
Subkeywords:
NB: The input PDB file must contain CRYST1 cards for the OUTPUT option to function.
The REPORT keyword controls the logfile output that the program generates.
Subkeywords:
(Optional) Specifies the end of keyworded input and starts AREAIMOL running.
Notes
For each area calculation performed by the program it will by default output an analysis of the accessible area by residue, by chain, and for the whole molecule. For each chain the accessible area of each residue will be listed, followed by the total for the chain. The reporting of areas per residue can be suppressed by using the REPORT RESAREA OFF keywords.
In the cases where only waters are considered (DIFFMODE WATERS, or MODEs HOH or HOHALL) an additional breakdown is presented of the waters which have no accessible area, and those which have areas < 5Å2, < 10Å2 and > 10Å2.
By default the program also outputs the contact area for each residue, chain and for the whole molecule. The contact area is defined as the area on the Van der Waals surface of an atom that can be contacted by a sphere of the given probe radius (see figure 2 above for a schematic representation of the contact area). The reporting of contact areas can be suppressed by using the REPORT CONTACT OFF keywords.
For modes NOHOH and ALL the program analyses the atoms which have been assigned accessible area and tries to determine how many isolated areas of surface there are (i.e. areas of surface which are unconnected to each other on the original molecule). Multiple isolated surfaces could represent any combination of:
For each isolated area of surface identified, the program reports the number of atoms, the total accessible area and the centre of mass.
In the case when differences in area are calculated (DIFFMODE other than OFF), an additional analysis is presented of the number of each atom type which have non-zero area differences. This is summarised in a table with the following quantities:
There is also a breakdown of accessible area differences by residue, chain and for the whole molecule.
Additional output can be obtained by specifying the VERBOSE keyword, which causes the program to print out diagnostic information such as recognised atom types and radii and the symmetry matrices derived from the symmetry cards. The REPORT keyword can also be used to select the desired output from the area calculations.
Analysis of surface accessible areas and area differences.
There were originally four programs to analyse solvent accessible area (AREAIMOL, RESAREA, WATERAREA and DIFFAREA). This version combines the function of the original set of programs into a single run which is controlled by the DIFFMODE keyword:
This mode analyses the accessible surface area of a molecule.
In the most basic mode of operation the program performs a single area calculation, obtaining the solvent accessibility of each atom under consideration. These individual areas are then used to obtain an analysis of the total accessible area for each residue, chain and for the whole molecule.
The MODE keyword can be used to exclude certain types of residue (e.g. waters) from the calculation. The effect of intermolecular contacts (which will reduce the accessible area) can be included using the SMODE keyword (which generates symmetry-related copies of the original molecule by applying the symmetry operations supplied with the SYMMETRY keyword) and the TRANS keyword (which will apply linear combinations of primitive lattice vectors to the symmetry-related molecules to generate further copies). Combining the primitive lattice vectors with spacegroup symmetry will effectively generate the crystal lattice.
This reproduces the function of the old AREAIMOL program followed by either WATERAREA or RESAREA as appropriate.
This mode compares the difference in accessible area due to the presence of intermolecular contacts, e.g. changes in accessible area due to oligomer formation.
Two area calculations are performed, one for each set of supplied symmetry operations (see SYMMETRY and TRANS keywords - if only one set of operators is supplied then the second set is assumed to consist of the identity). The difference in accessible area on each atom is then calculated and the overall area differences analysed.
The SMODE keyword has no function under the DIFFMODE IMOL option, and the SYMMETRY keyword can appear twice: each occurrence gives the operators for one calculation of accessible area. Other keywords maintain their function and take effect during both calculations.
This mode only considers waters and compares the difference in accessible area when waters are treated as solvent as opposed to as protein (i.e. water treated as protein can 'obscure' surface area on other waters).
Only one set of coordinates is input, and two separate area calculations are carried out (the first treating waters as solvent, i.e. equivalent to MODE HOH, and the second treating them as protein, i.e. equivalent to MODE HOHALL). The area differences are then calculated and output.
The results of the calculations can be interpreted as follows:
The value of the area difference for each water listed is equal to the reduction in accessible area due to being obscured by neighbouring waters. Waters buried completely in protein will not be listed in the area difference analysis.
The MODE keyword has no function under this option, although the other keywords maintain their function and take effect during both calculations.
This mode compares the difference in accessible areas for two similar molecules, e.g. changes due to substrate or ligand binding.
Two input coordinate files are required, and two separate area calculations are carried out, one for each set of coordinates. The same MODE and symmetry operators etc (if relevant) are used in each case, so the resulting area differences will depend only on differences between the contents of the files. Area differences are calculated only for those atoms which are common to both files.
E.g. if one file describes a protein bound to a ligand and the other describes the protein alone, then using this mode will calculate the change in surface area of the protein in the presence of the ligand, or more specifically the area obscured by the ligand.
The following comments are based on those in the original documentation:
The area calculations also depend critically upon various parameters, such as the probe radius (taken to be 1.4Å for most calculations) and the van der Waals radii chosen for different atoms. Many programs (including AREAIMOL) choose one radius for all carbons, one radius for all nitrogens, one for all oxygens, whereas others (e.g. SURFACE) are able to differentiate between different carbons (aliphatic, aromatic etc.), different nitrogens and so on.
SURFACE assigns the Van der Waals radius for a given atom according to both the element and also the residue in which it appears, and thus may lead to differences in estimates of the accessible area.
Note that SURFACE calculates both the accessible area and the contact area, but does not include options for accounting for intermolecular contacts.
One of the factors limiting the accuracy of the AREAIMOL calculation is the algorithm used to divide up the surface of each expanded Van der Waals atom. As of CCP4 6.1, Ian Tickle has implemented an improved algorithm which divides the surface up more evenly according to a "spiral" pattern based on Saff and Kuijlaars (1997). Using this improved algorithm the calculated values of the ASA appear to be more accurate and stable over a wide range of PNTDEN values.
The maximum size of protein that the program can handle is hardcoded in the parameter MAXNET in the source code file areaimol.f:
PARAMETER (MAXNET=6000000)
There is not a straightforward relationship between the number of atoms in the structure and the minimum size of this parameter however the program will detect when it is too small and will report what the required value is before terminating, for example:
ERROR: Dimension of NET too small in program. Need: 12362350 Currently: 6000000 AREAIMOL: Parameter MAXNET too small
In this case it should be sufficient to edit the value of the MAXNET parameter to be at least the recommended size (i.e. 12362350 in this example) and then recompile the program.
The number of symmetry operations that the program can handle is hardcoded in the parameter MAXSYM in the source code file areaimol.f:
PARAMETER (MAXSYM=60)
In the event that the program stops with the message Too many symmetry operations, this limit has been exceeded. The program will suggest a new value for this parameter - you will need to update all occurrences before recompiling the program.
Originator: Peter Brick, Imperial College
Substantial modifications/additional features: Peter Briggs, Ian Tickle
surface, contact, CCP4 Newsletter article about SURFACE and AREAIMOL